

Abstract
Background: The first two autologous CD19 chimeric antigen receptor T (CAR T) cells targeting CD19 have now been approved for the treatment of ALL and refractory lymphomas. Despite impressive responses in these diseases, results remain inconsistent in chronic lymphocytic leukaemia (CLL). It is unknown if this reflects CAR design or an effect of the underlying function of CLL T cells. These 2nd generation CAR T cells require CD28 or 41BB co-stimulatory signalling domains, but these have not been compared directly in humans. Pre-clinical models afford the opportunity to do this, however, modelling of CAR T cells has mostly been performed in vitro or using immunodeficient mice, limiting the ability to study more complex immune biology. CLL is associated with a tumour supportive microenvironment and T cells exhibit multiple functional defects and features of exhaustion. These T cell defects in CLL are closely recapitulated in Eμ-TCL1 (TCL1) mice, and induced in healthy mice by adoptive transfer (AT) of murine CLL cells. We aimed to demonstrate the effect of CLL T cell dysfunction on CAR T cell efficacy and compare CD28 and 41BB directly.
Methods: Immunocompetent C57BL/6 mice (WT) received AT of pooled 20 x106 TCL1 cells from fully leukemic TCL1 mice from the same background. Syngeneic donor CAR T cells were either pooled spleens from WT mice or WT mice given AT CLL with CLL load >80%. Both groups were aged matched (approx. 3 months). Splenoctyes were enriched for CD3+ with magnetic beads then activated with anti CD3/CD28 Dynabeads (Thermofisher) and rIL-2 (Roche). They were transduced with retroviral supernatant from either SFG-m19BBmZ-GFP (CD19-41BB) or MSGV-1D3-28Z-1.3mut (CD19-CD28) and cultured for 4 days when they were injected into 48 mice in total. Mice were given 100mg/kg intraperitoneal cyclophosphamide on D-1 followed by 6-8 x106 CAR T cells (or untransduced T cells). Mice were bled weekly to assess CLL load and T cell subsets and were culled when they appeared sick or peripheral blood (PB) CLL>70%.
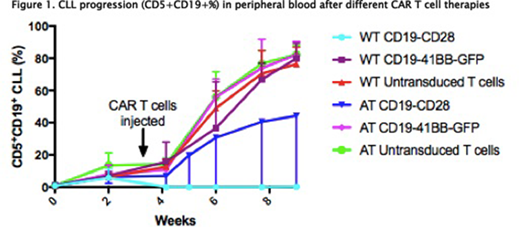
Results: CAR T cells derived from WT and AT T cells exhibit different phenotypes. WT CAR T cells proliferate more readily in culture and exhibit significantly higher transduction efficiencies in the CD8 subset although CD4 transduction is preserved. Following activation and transduction WT CAR T cells have a CD4: CD8 ratio of 1:1 whilst those from AT are heavily skewed to CD8. In both groups >90% T cells are CD44+. PD1+ expression in both CD4 and CD8 subsets is significantly higher in AT compared to WT CAR T cells. Mice treated with the CD19 -41BB CAR derived from WT and AT T cells or untransduced T cells did not respond, whereas 100% of mice treated with CD19-CD28 CAR derived from WT T cells had a complete response with loss of normal B cells 1 week post CAR T cells injection compared to 50% of mice treated with CD19-CD28 from AT T cells. All non-responding mice were culled at week 8 due to progressive leukaemia as were control mice treated with untransduced T cells. All mice with an established response had a continued complete response for 5 weeks following CAR T cell injection. Half of these mice were culled for phenotypic comparison and the other half observed for survival analysis. Those mice that responded and culled at week 8 had equal spleen size (0.1g) to age matched WT mice controls whilst non-responding mice had significantly larger spleens (0.5-3.3g). CAR T cells were only detectable in the PB +1 week post injection. In the PB there was restoration of CD4: CD8 ratios in responding mice compared to leukemic mice. PD1 expression in the spleen and bone marrow in CD3+CD8+ and CD4+ T cells normalised in responding mice compared to non-responding mice.
Conclusion: AT of TCL1 CLL into immunocompetent mice is a viable model to study in vivo CAR T cell function and the host immune response. CAR T cells derived from WT T cells lead to a complete response in all of the mice but this response is significantly reduced if T cells exposed to CLL are used. Time to relapse for these responding mice has not been reached. We postulate that failure of the CD19 -41BB CAR in vivo relates to rejection of the GFP construct. There are significant differences in PD1 expression between WT and AT derived CAR T cells, which suggest strategies to repair exhausted T cells may improve the clinical response to CAR T cells in CLL. This provides the rationale for our on going studies of PD1/PDL1 blocking drugs in combination with CAR T cells in this immunocompetent pre-clinical model.
Gribben:Medical Research Council: Research Funding; Celgene: Consultancy, Honoraria, Research Funding; Acerta Pharma: Honoraria, Research Funding; Janssen: Honoraria, Research Funding; Novartis: Honoraria; Pharmacyclics: Honoraria; NIH: Research Funding; Kite: Honoraria; TG Therapeutics: Honoraria; Wellcome Trust: Research Funding; Cancer Research UK: Research Funding; Unum: Equity Ownership; Roche: Honoraria; Abbvie: Honoraria.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal